2025年9月28日, George Church(哈佛教授,基因组学大佬,搞出蛋白条形码的那个)创业的Manifold Bio公司
发表了一篇《mBER: Controllable de novo antibody design with million-scale experimental screening》.这篇文章总体思路是很典型的后BinderCraft作品,最大的一个亮点是同当初的Chai Discovery那样, 在大量的靶点上拿到了Hit.那么究竟如何,让我们细细阅读分析.
代码仓库: GitHub - manifoldbio/mber-open
开源许可: MIT
背景
幻觉设计(Hallucination)是蛋白设计领域的一种经典技术方案, 其核心思想是: 通过特定优化目标和方式,去优化一个随机初始化的序列, 使其能够折叠成设计目标.具体的优化过程可以是像基于梯度的优化, 也可以是MCMC采样的优化.
而alphafold2的成功, 让基于结构模型梯度的优化成为可能, 比如colabdesign就是基于af模型做优化设计的一个通用框架,再次基础上衍生出了BindCraft、FoldCraft和Germinal等具体方案.然而这些方案多数都在抗体设计的性能上表现不佳, 主要原因是抗体结构预测对AF2是很大的挑战, 且其优化的序列在表达设计上较为困难.
本文的核心点
- 引入ESM2等LLM对优化过程概率进行偏好引导
- 引入抗体结构、抗原结构的独立模板, 提升结构的可靠性
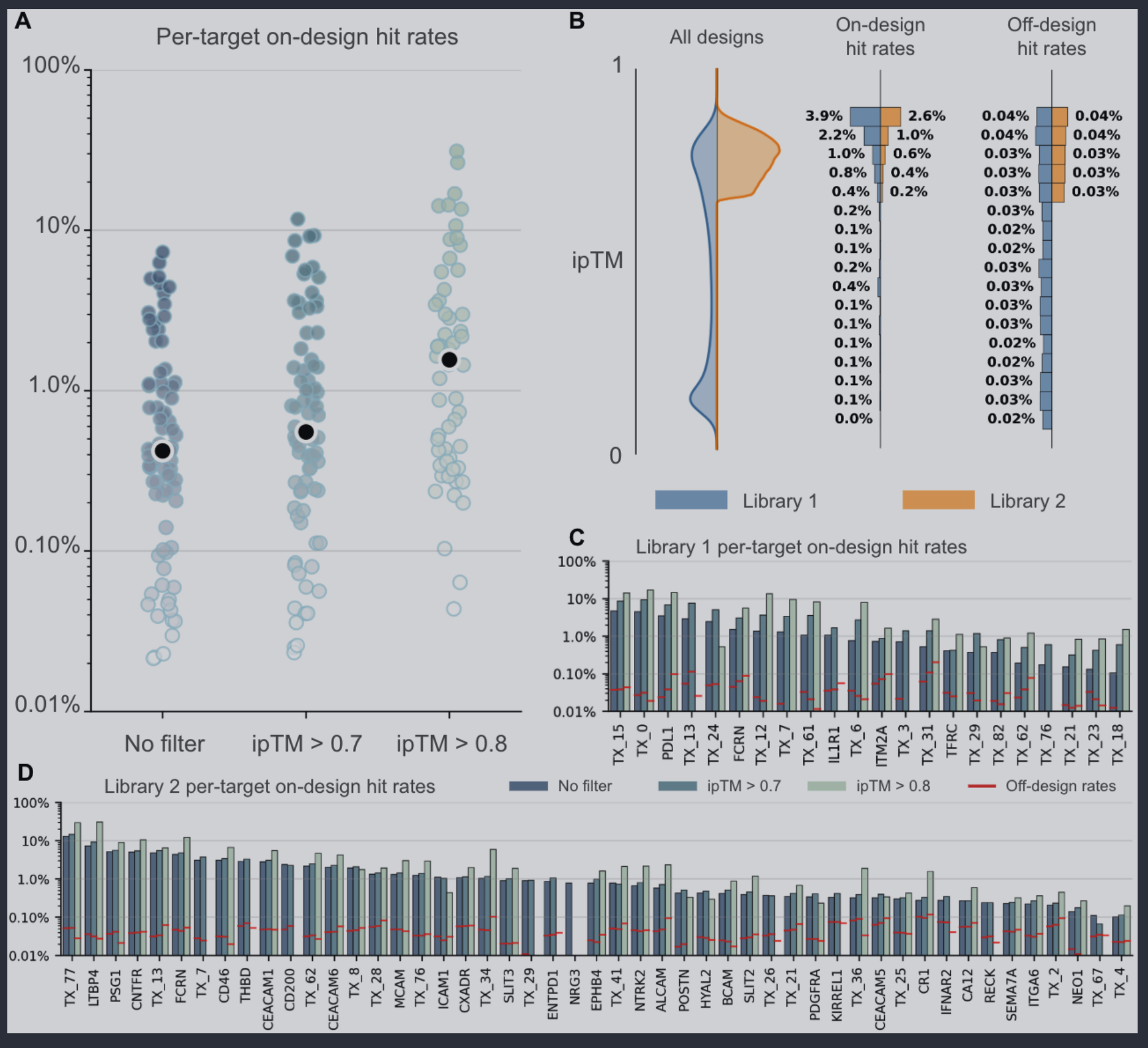
- ipTM在筛选上展现较好的相关性
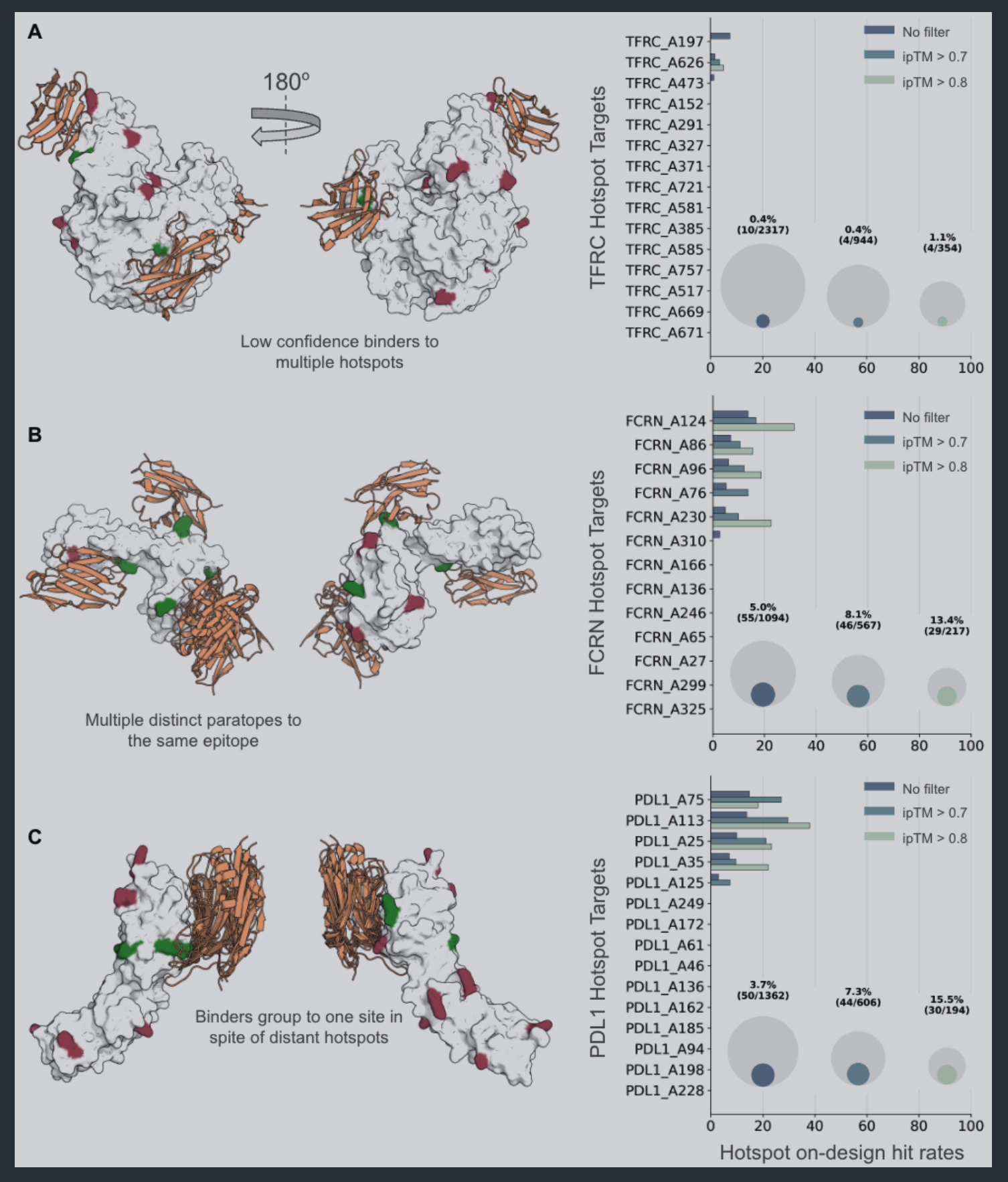
- 针对436个靶点构建了两个phage库的百万级VHH样本, 并在145个靶点上拿到binder.
算法架构解析
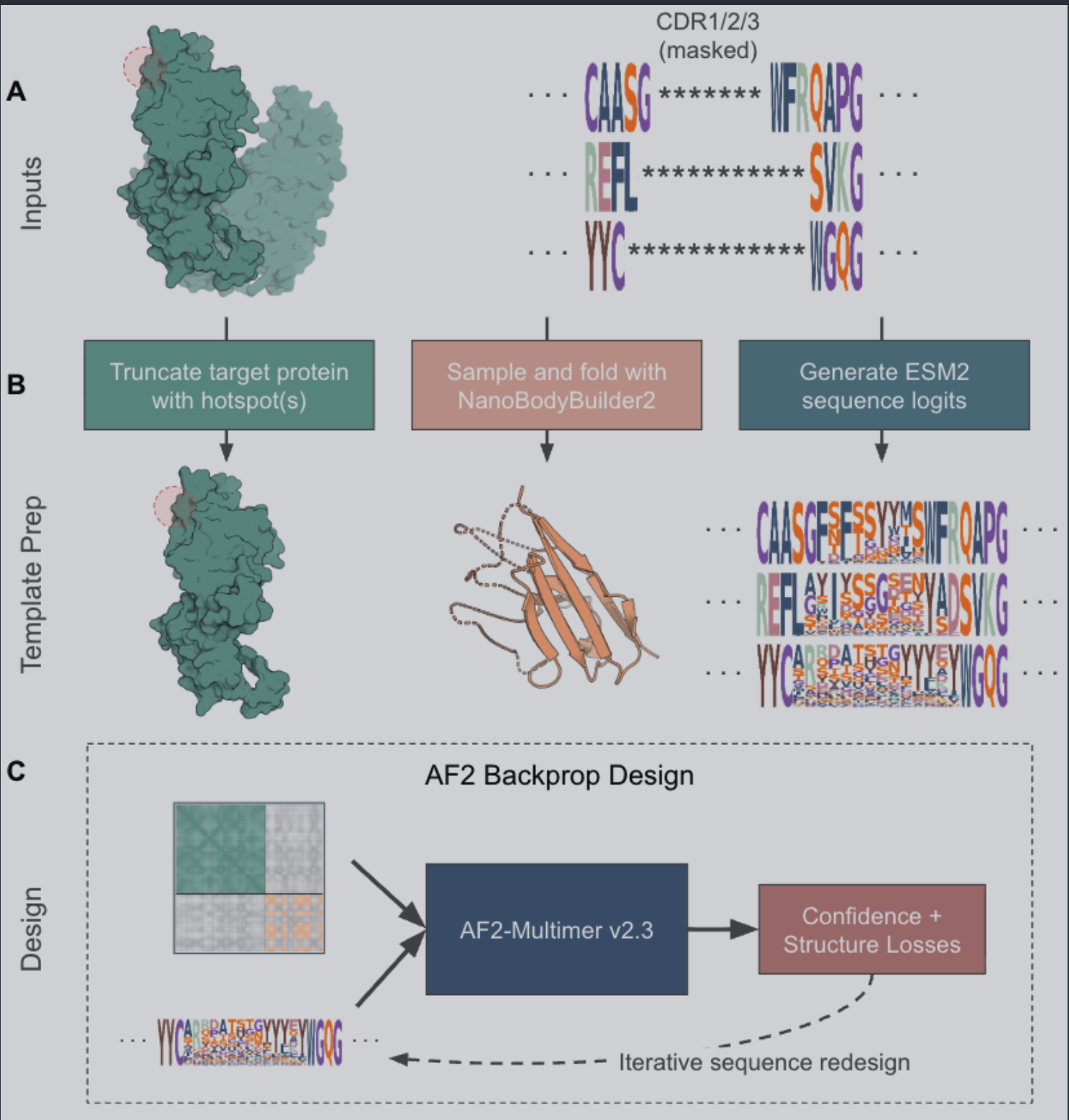
如其文章所讲述, 算法分为4个阶段: 输入准备、模板构建、序列生成、序列评估.
输入准备
主要包括抗原结构的准备、表位关键残基(hotspot)的选择以及VHH骨架germline的挑选.如后文所说,
表位hotspot的挑选对设计结果影响很明显, 但可惜文章并没能解决这个选择困难问题.但这个算法支持了
对target结构的裁切, 从而减少体系大小, 提高计算性能.而VHH的germline本文都选了IGHV3-23这个
最经典的.
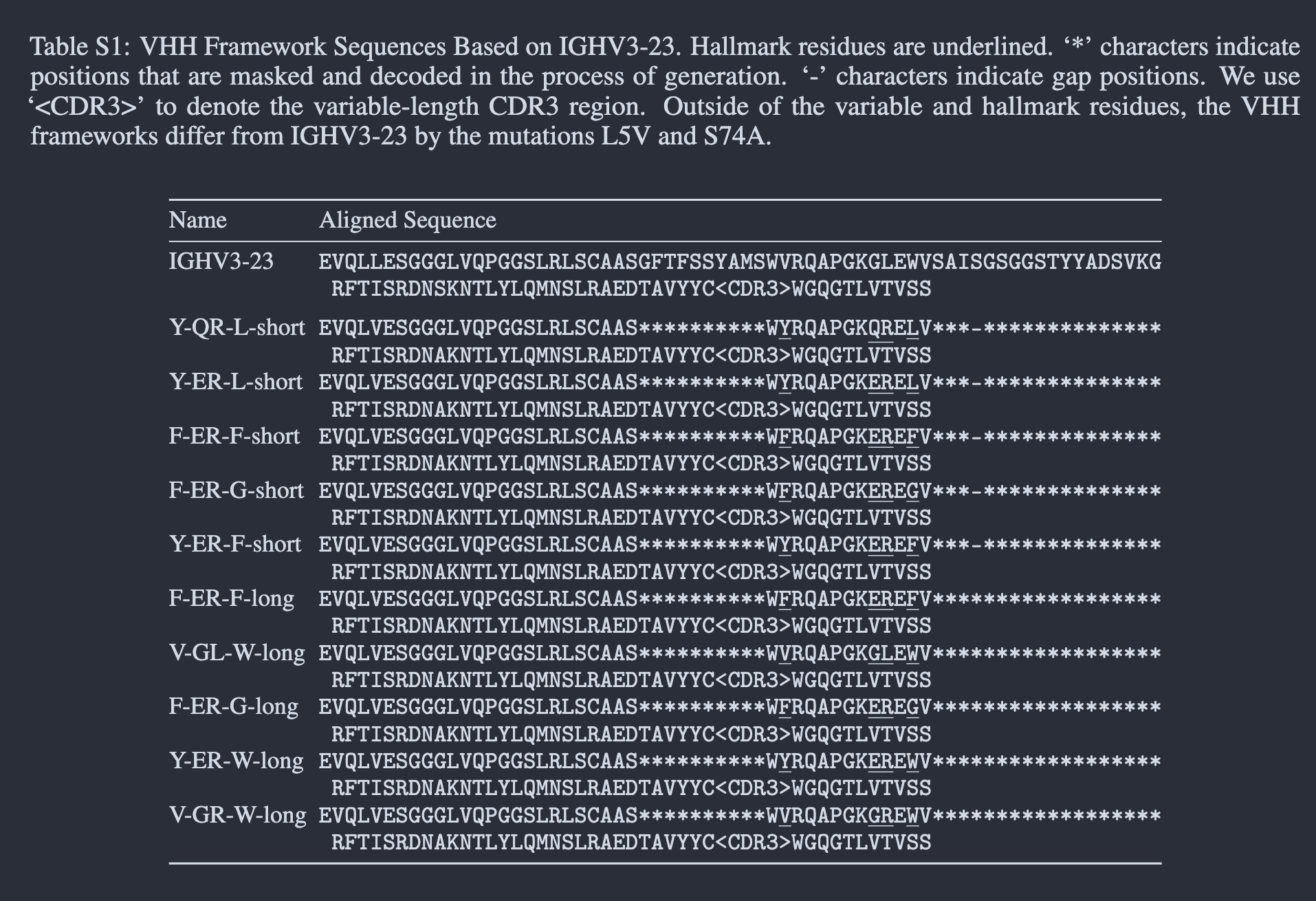
关于具体的truncated蛋白过程, 作者是下列step:
- 从AFDB fetch结构
- 使用TMbed预测胞外domain并truncated
- 根据SASA, 选择至少10个表面残基作为hotspot
- truncated掉25angstrom外的残基.
但我个人给出一些concern:
- 如果有晶体结构的需要AFDB吗?
- 如果Uniprot记载了细胞外domain, 还需要TMbed吗?
- 表位hotspot之间是否有额外空间要求?
- 基于hotspot周围25埃的truncated, 是否需要保障序列的连续性?
模板的准备
此前, BindCraft等流程没有直接构建适合抗体的结构模板, 而本文通过利用ESM2对VHH mask区域进行随机采样,
并使用NanoBodyBuilder2建模, 从而构建了抗体结构模板.这个模板会和抗原结构放在一个pdb中.但在设计时它们之间
的pose并不会被参考.
colabdesign流程的调整
与BindCraft不同, mBER总体是使用design_3stage的colabdesign protocol进行幻觉设计的,比4stage BindCraft少了一步proteinMPNN优化.同时为了提升抗体-抗原复合物建模,这里修改支持了对抗原结构和抗体结构输入的独立template, 并掩蔽了inter-chain contact map, 故而而不会参考其随机的pose.
当然最核心的调整是增加了LLM的Sequence bias, 具体通过一个模拟退火过程来融合:
其中$\lambdat$是会随着设计步数逐渐增加, 而$T_t$是随着步数下降的退火温度, $l{design}$是直接用AF2模型反向传播优化的序列logits, $l{bias}$是由ESM2前面提供的bias logits, $T{bias}$则是bias logits的温度(没有说明这里是否有退火).
loss项与BindCraft较为相似, 以cross chain pAE为主, 辅以其他af2的confidence metrics.
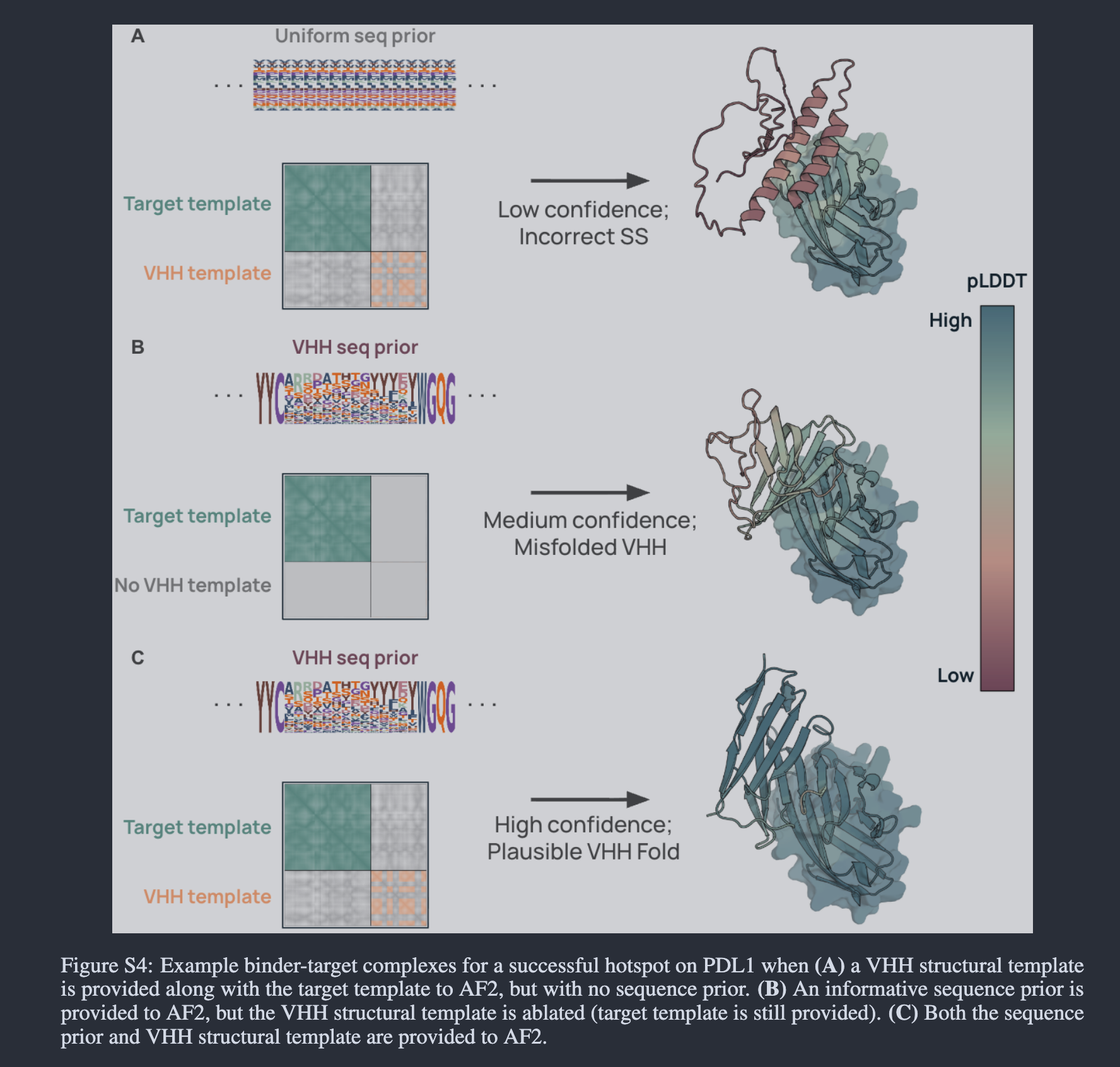
结果的评估
此外, mBER每次都以4/5的AFM权重进行design, 剩余一个weight用来做评估, 设置与design一致, 均为3个recycle(似乎会启用amber relax).
phage文库的构建与结果
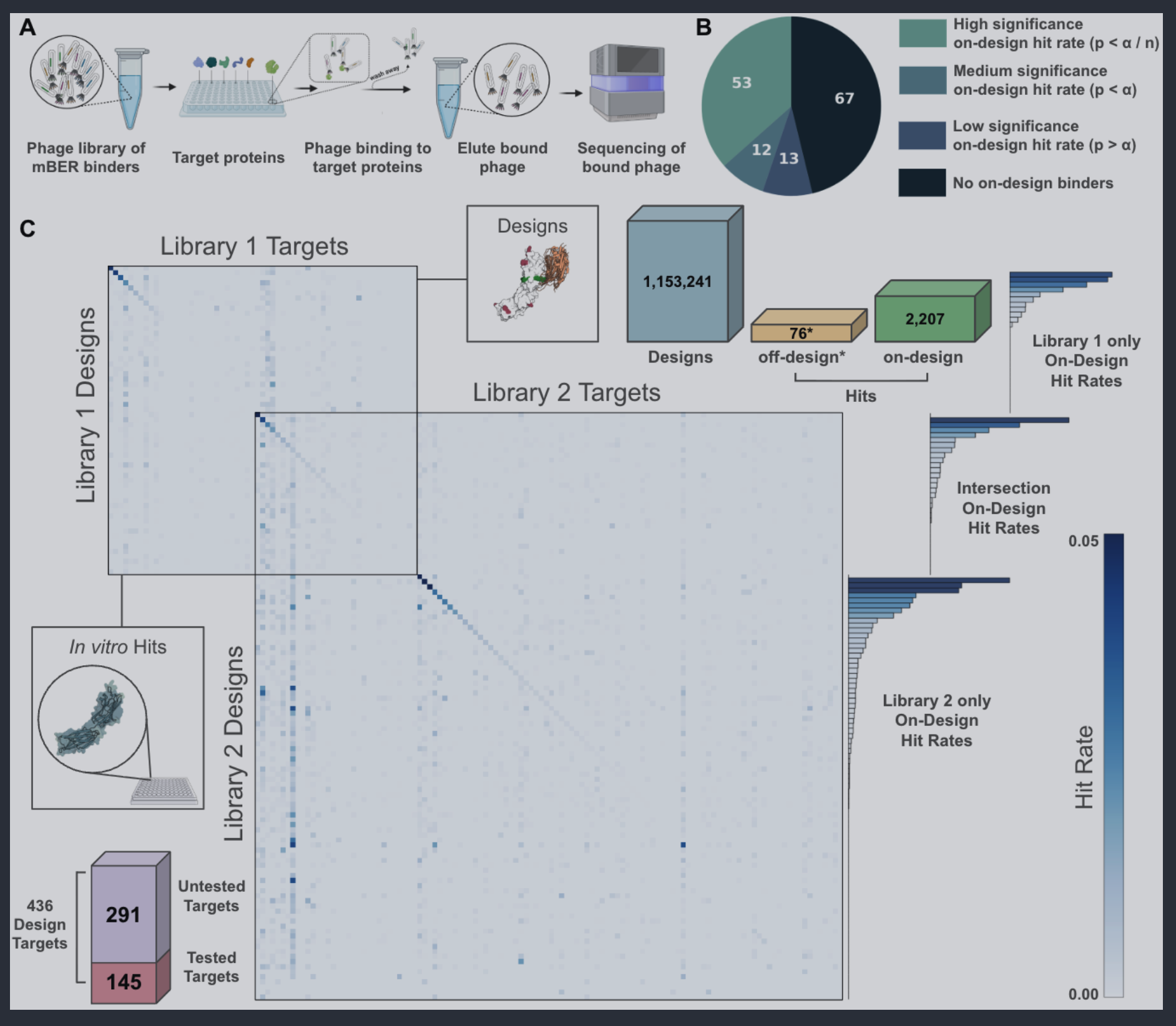
libary1
libary2
表位设计的成功率