论文:Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
来源:ICLR 2026
机构:NVIDIA
官网:https://research.nvidia.com/labs/genair/proteina-complexa
代码:https://github.com/NVIDIA-Digital-Bio/proteina-complexa
论文评审:https://openreview.net/forum?id=qmCpJtFZra
一句话总结
Proteina-Complexa 是 NVIDIA 提出的首个统一生成式预训练与推理时间优化的蛋白质结合剂(binder)设计框架,结合了生成模型与”幻觉”(hallucination)方法的优势,在蛋白质靶点、小分子靶点和酶设计任务上均达到了新的 SOTA 水平。
核心创新点
Proteı́na-Complexa 的核心贡献可提炼为以下三点:
- 统一范式(Unified Paradigm): 首次打破了“单纯生成式模型(如 RFDiffusion)”与“单纯幻觉优化(如 BindCraft)”的二元对立,提出了 “潜空间生成搜索(Latent Generative Search)”。它将强大的生成先验与推理端优化相结合,利用生成模型作为高效的搜索起点。
- Teddymer 数据集: 针对 PDB 中高质量复合体数据稀缺的瓶颈,研究团队从 AlphaFold Database (AFDB) 的 47M 结构中提取“域-域(Domain-domain)”相互作用,合成了规模达 3.5M 聚类中心 的 Teddymer 数据集,为全原子预训练提供了规模化支撑。
- 推理端缩放(Test-time Scaling): 借鉴 LLM 的 Reasoning 思路,在采样过程中引入 Feynman–Kac Steering (FKS)、MCTS 及 Beam Search。通过投入更多推理算力,模型能显著提升在复杂靶点上的设计成功率,实现性能随算力投入的非线性增长。
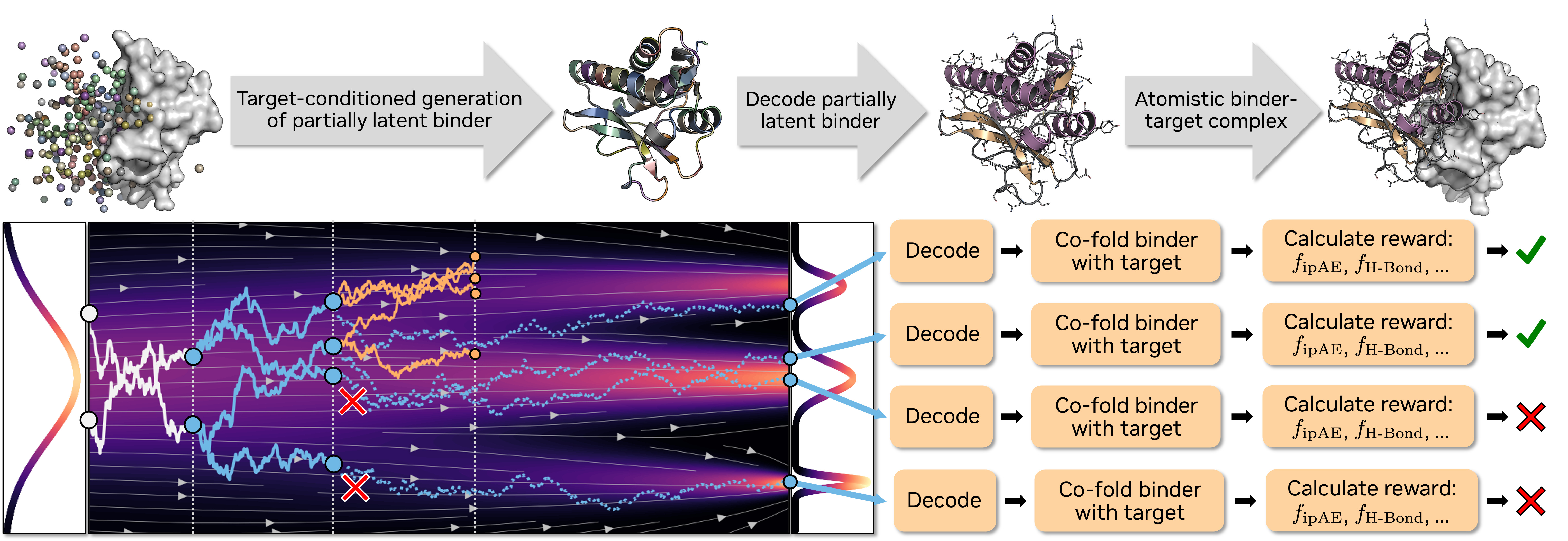
1. 统一生成式与幻觉方法:基于 La-Proteı́na 的部分潜空间流匹配
架构设计原理解析
在处理全原子数据时,计算复杂度是首要挑战。Complexa 采用了“部分潜空间”架构:将 $C\alpha$ 骨架保持在显空间以维持几何直观性,而将复杂的侧链分布与氨基酸序列压缩至 8 维连续潜变量中。这种设计的关键在于 VAE 模块的冻结共享策略:VAE 仅在单体 Binder 上预训练且在后续流程中保持冻结,而由 Transformer Denoiser 负责处理复杂的靶点条件化逻辑,实现了计算效率与表达精度的平衡。
算法核心组件表
| 组件名称 | 输入/输出内容 | 在 Complexa 中的关键作用 |
|---|---|---|
| VAE Encoder/Decoder | 全原子坐标 $\leftrightarrow$ 潜变量 + $C\alpha$ 坐标 | 降低全原子数据的维度,将混合的离散/连续空间转化为统一的连续向量表示。 |
| Transformer Denoiser | 噪声潜变量 + 目标特征 | 利用 Multi-head Pair-biased Attention 建模,预测向量场。避免了昂贵的三角形更新层。 |
| 条件化机制 | Hotspot 标记 + 目标 Atom37 | 采用潜空间条件化机制,通过跨注意力引导 Binder 的全局定位与界面匹配。 |
该架构摒弃了传统模型中沉重的计算模块,单次采样速度极快(约 15.6 秒),这使得大规模的搜索策略(如 MCTS)在工业应用中变得可行。
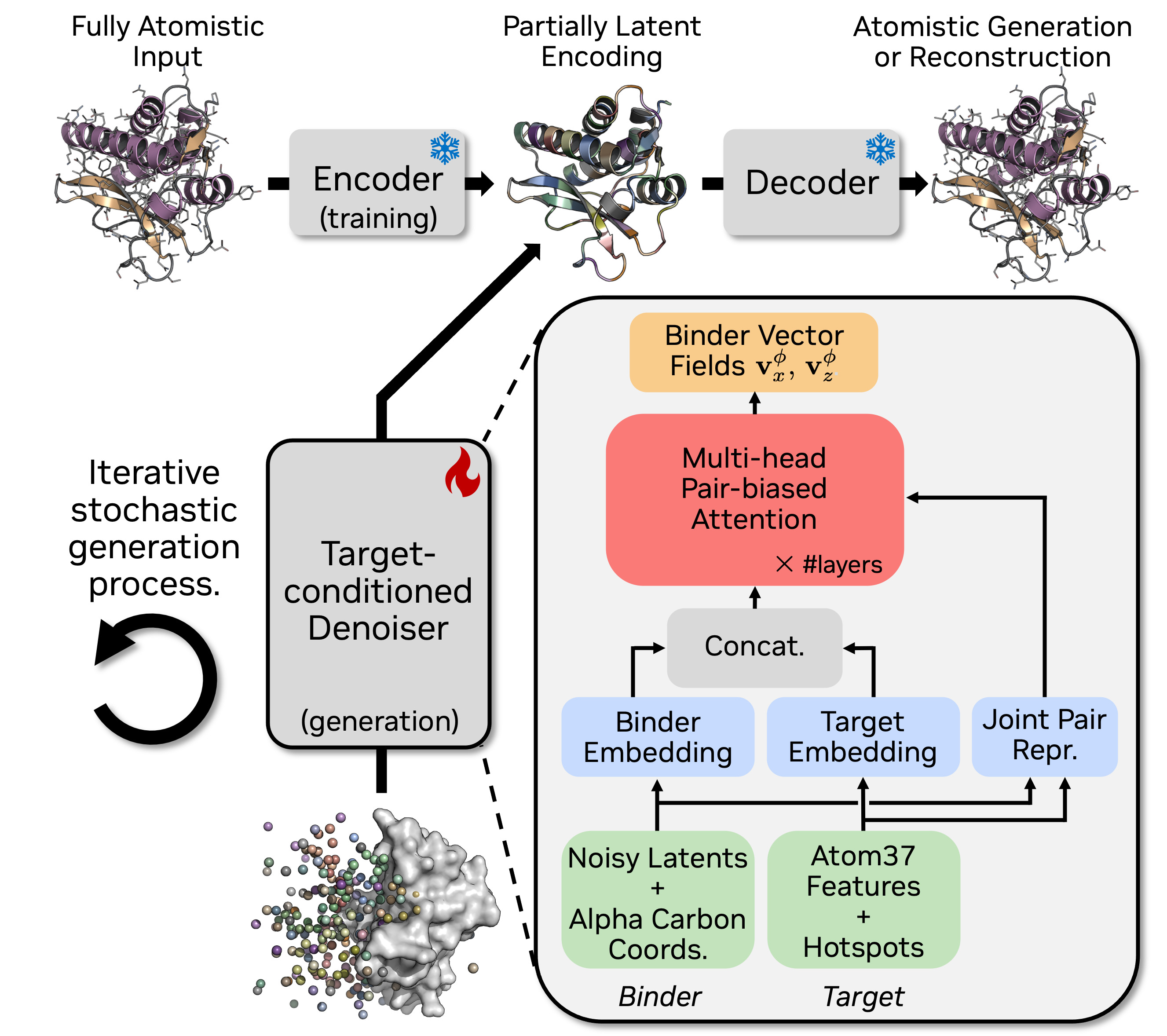
图注:A frozen autoencoder (top) encodes fully atomistic proteins into a partially latent representation (alpha-carbon coordinates + continuous per-residue latents) and decodes them back. The target-conditioned denoiser (bottom) concatenates embedded target features (Atom37 coordinates, amino acid identity, hotspot tokens) with the binder’s noisy latent and backbone embeddings, processing them jointly through multi-head pair-biased attention layers.
2. 数据集的收集与处理:Teddymer 与多阶段策略
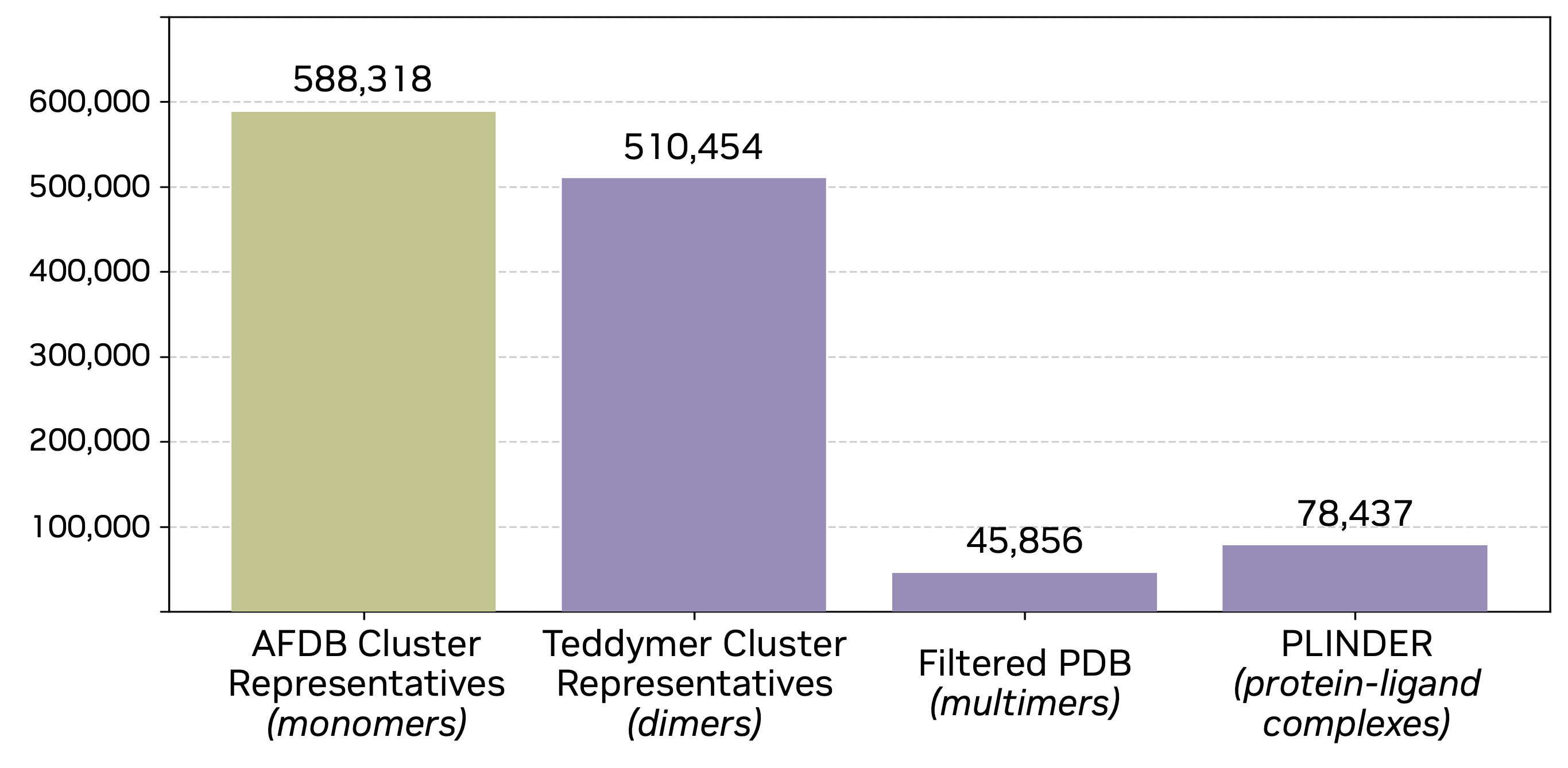
模型训练用到了下列四个数据, 考虑由于 PDB 中实验解析的复合物结构有限,作者提出从 AlphaFold Database (AFDB) 中提取蛋白质结构域相互作用,构建大规模合成 binder-target 数据集(Teddymer):
数据质量直接决定了生成模型的“物理直觉”。PDB 中约 22.5 万个条目对于全原子生成而言过于稀疏。Complexa 团队提出了一个深刻的洞察:单体蛋白内部域与域之间的界面物理规律,与链间界面高度一致。
- 数据源: 从 AFDB50 数据库中提取 47M 具有 TED(Encyclopedia of Domains)标注的预测结构。
- 合成逻辑: 将单体蛋白按 TED 标注拆分为独立域,模拟为“结合剂-靶点”对。
- 筛选标准: 仅保留符合 C.A.T. 标注、界面 pLDDT > 70 且 ipAE < 10 的高质量二聚体。
- 规模: 从 10M 原始二聚体中,通过 Foldseek 聚类生成 3.5M 聚类中心,最终精炼出 510k 高质量训练集。
为了证明模型学习的是物理通用性而非简单的结构记忆,团队使用了 Foldseek 进行结构层级的聚类去重。通过严格过滤测试集靶点的同源结构,确保模型在处理新靶点时依靠的是从 Teddymer 中汲取的界面相互作用规律。
3. 推理时间优化策略
借鉴大语言模型推理时扩展技术,引入多种搜索策略:
- Best-of-N:采样多个候选,选最优
- Beam Search:束搜索引导生成
- Feynman-Kac Steering:基于能量的引导
- MCTS:蒙特卡洛树搜索
测试的靶点与结果
In silico Performance and Benchmarking
Generative Base Model
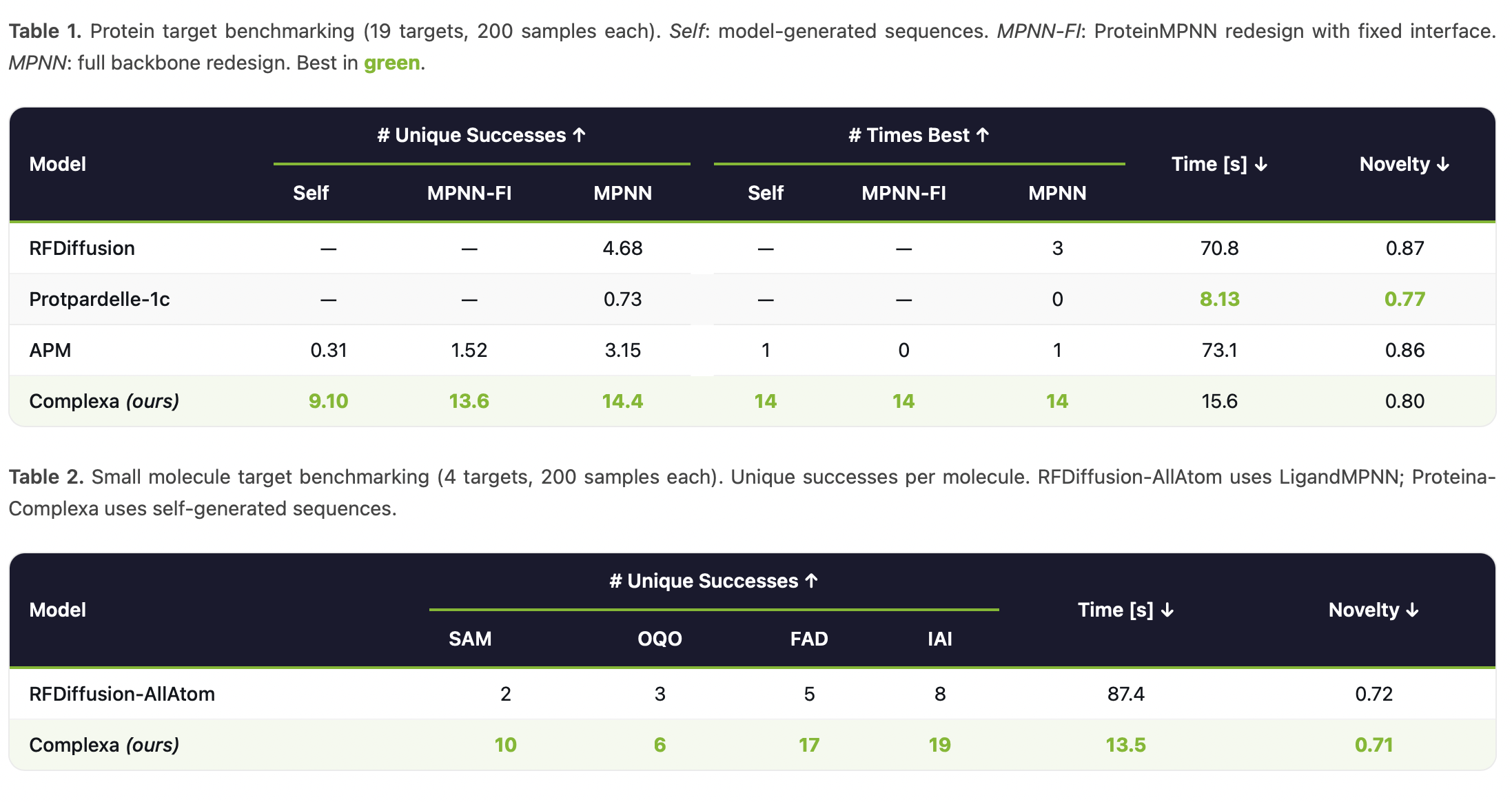
Inference-Time Compute Scaling
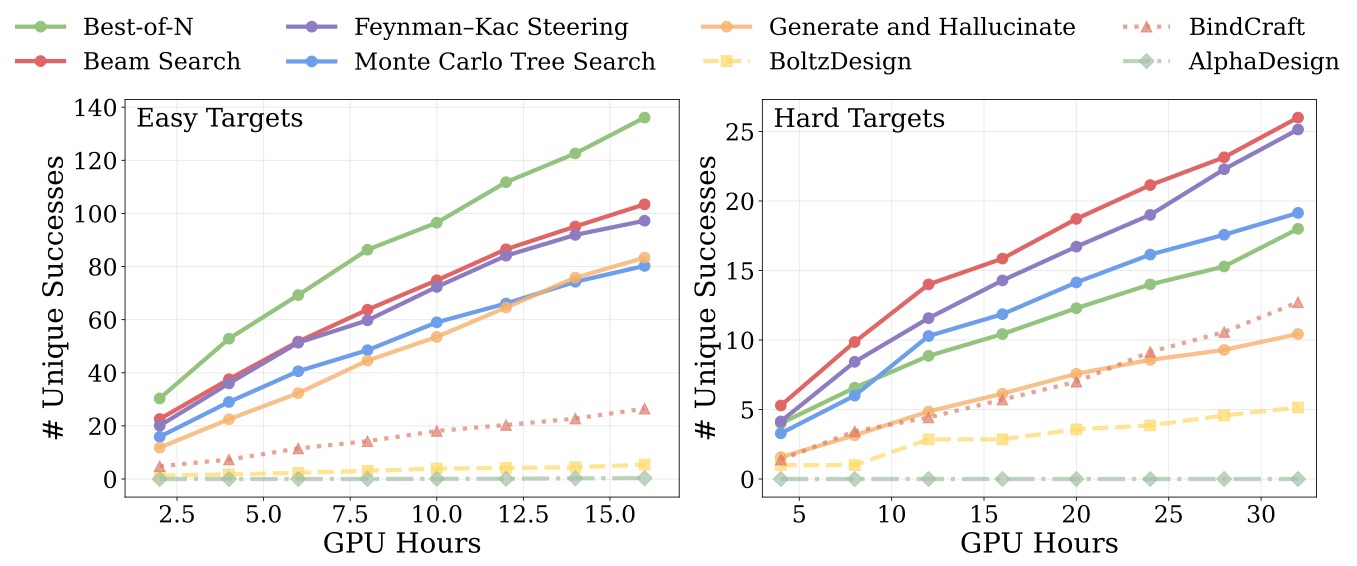
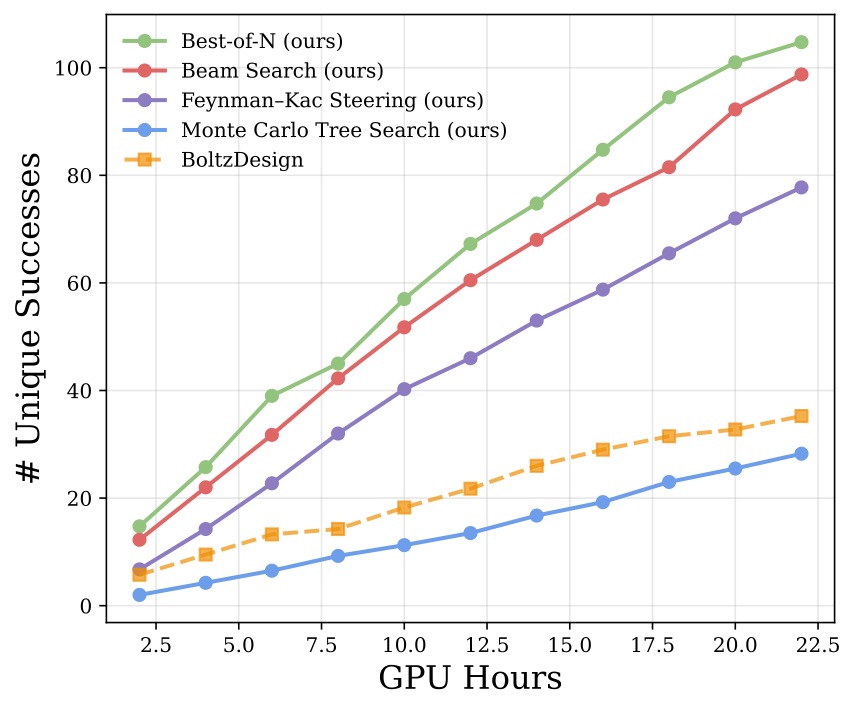
Unique success rate vs. optimization time (GPU hours) for easy targets (left) and hard targets (right). Proteina-Complexa’s search methods (colored curves) consistently outperform hallucination baselines (BindCraft, BoltzDesign, AlphaDesign) under normalized compute budgets.
- 蛋白质靶点
测试了多个具有挑战性的蛋白质靶点:
- TNF-α(三链靶点)
- Claudin-1(跨膜蛋白)
- IFNAR2(干扰素受体)
- IL-17A(双链靶点)
- PD-1/PD-L1 等
- 小分子靶点
- SAM、IAI、FAD、OQO 等
酶设计任务 (AME Benchmark)
在 Atomic Motif Enzyme (AME) 基准上测试,包含 41 个任务。
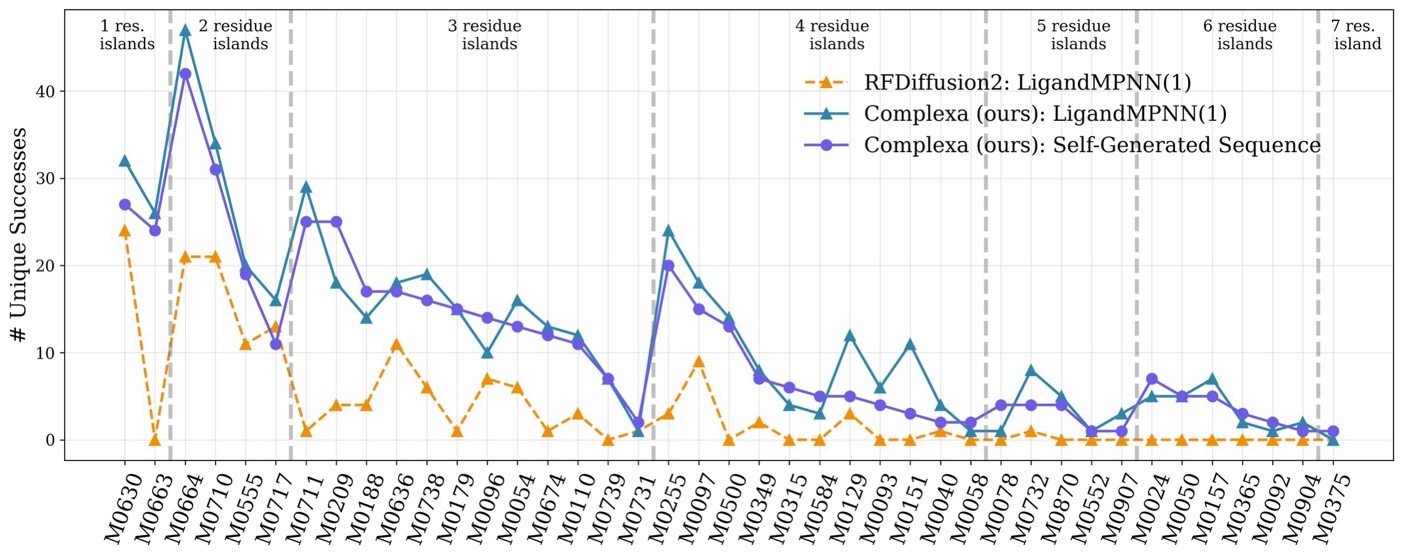
Number of unique successes per task (41 tasks, 100 samples each) for Proteina-Complexa vs. RFDiffusion2, comparing self-generated sequences, single LigandMPNN redesign, and best-of-8 LigandMPNN redesigns. Proteina-Complexa outperforms RFDiffusion2 on the vast majority of tasks across all evaluation settings.
成功率对比
| 方法 | 蛋白质靶点 | 小分子靶点 | AME 基准 |
|---|---|---|---|
| RFDiffusion | 基准 | - | 基准 |
| Protpardelle | 较低 | - | - |
| BindCraft (幻觉方法) | 基准 | - | - |
| Proteina-Complexa | 最高 | 最高 | 最高 |
关键发现:
- Complexa 在 38/41 个 AME 任务上优于 RFDiffusion2
- 推理时间优化策略在归一化计算预算下显著优于幻觉方法
- 无需序列重设计(self-sequence 即可达到高成功率)
湿实验证
本文的一个亮点是进行了 wet lab 验证!据官网报道,Proteina-Complexa 设计的 binder 已在实验中获得验证,显示 in-silico 成功可以转化为实际结合活性。
详细实验数据见:验证实验论文
生成示例
官网提供了多个生成示例的3D可视化, 大分子
代码开源情况
许可证
根据 GitHub 仓库,代码采用 NVIDIA Software License(非标准开源许可证),需遵守 NVIDIA 的许可条款。
部署方式
提供两种部署方式:
- 本地部署:Ubuntu 22.04+,需要构建 UV 环境
- Docker 容器:推荐方式,包含所有依赖
1 | # 克隆仓库 |
模型权重
需要从 NVIDIA NGC 下载模型权重:
- 蛋白质 binder 模型
- 小分子 binder 模型
- AME(motif scaffolding)模型
1 | # 初始化 |
依赖工具
- AlphaFold2 / RoseTTAFold3(用于 reward 评估)
- Foldseek、MMseqs2、DSSP、SC 等生物信息学工具
训练策略
阶段性训练
- 第一阶段:仅在 monomer 上训练,获得通用蛋白结构生成能力
- 第二阶段:在 Teddymer + PDB multimers 上训练 binder-target 对
- 第三阶段:高质量实验多聚体精调
损失函数
- 部分潜在流匹配损失
- 引入 Translation Noise(平移噪声)增强 binder 定位能力
消融实验表明:无 Translation Noise 会导致 binder 放置位置不佳;无 Teddymer 数据会导致性能大幅下降。
优缺点分析
todo
值得探索的方向
- DNA/RNA 靶点:扩展到核酸靶点
- 联合训练:单一模型支持多种分子模态
- 更多实验验证:更多靶点的 wet lab 验证
- 特异性/热稳定性:集成更多评估指标
参考文献
- Didi, K. et al. (2026). Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute. ICLR 2026.
- Geffner, T. et al. (2026). La-Proteína: Fully Atomistic Protein Generation with Flow Matching.
- Watson, J. et al. (2023). RFDiffusion: Protein binder design.
- Pacesa, M. et al. (2025). BindCraft: Hallucination-based binder design.